
Czym są geny i chromosomy?
Geny są unikalnym zestawem instrukcji wewnątrz każdej komórki organizmu i decydują o przekazywaniu cech potomstwu. Geny warunkują dziedziczenie takich cech jak np. kolor oczu, włosów ale też np. odporność czy podatność na daną chorobę. Geny kontrolują także budowę poszczególnych komórek tworzących poszczególne narządy.
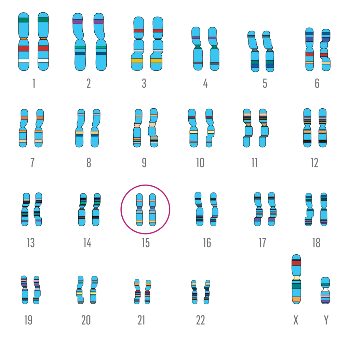
Pod względem fizycznym geny są liniowymi odcinkami DNA, które w każdej komórce zlokalizowane jest w jądrze komórkowym. Człowiek posiada w każdej komórce 46 długich cząsteczek DNA, każda z tych cząsteczek, podczas podziału komórki, tworzy jeden chromosom. Każdy chromosom zawiera tysiące genów. W organizmie ludzkim pogrupowane są one w 23 pary, jak na rysunku poniżej. Zazwyczaj dziedziczymy jeden chromosom z pary od mamy a drugi od taty.
W celu uwidocznienia w mikroskopie świetlnym chromosomów w komórce, wykonuje się badanie cytogenetyczne, czyli badanie kariotypu. Dzięki zastosowania odpowiednich odczynników, chromosomy można rozróżnić dzięki wzorowi prążkowemu oraz ich różnej długości.
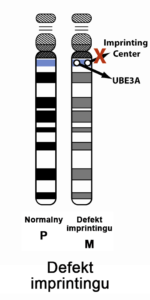
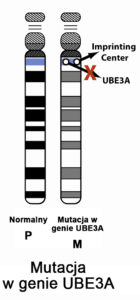
Niektóre geny działają inaczej, w zależności od tego, czy zostały odziedziczone od mamy, czy od taty – tak dzieje się w przypadku genu, który odpowiada za zespół Angelmana – aktywną kopią jest tylko ta, pochodząca od mamy. Taką sytuację nazywamy rodzicielskim piętnem genomowym (albo imprintingiem). Różnice w działaniu genów matczynych i ojcowskich wynikają z pewnych zmian chemicznych dotyczących samych genów (a dokładnie ich metylacji). Gen zmetylowany (w przypadku Zespołu Angelmana – pochodzący od ojca) jest nieaktywny.
Jak powstaje zespół Angelmana?
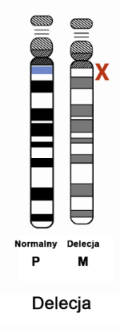
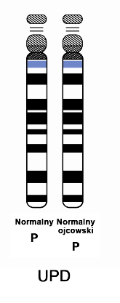
Objawy zespołu Angelmana wynikają z zakłócenia funkcji genu UBE3A, kodującego enzym – ligazę ubikwitynową. Gen ten jest zlokalizowany na chromosomie 15 w regionie q11.2-q13. Wyróżnia się 4 główne mechanizmy, które zakłócają funkcję tego genu, a przez to uważane są za podłoże zespołu Angelmana:
Poradnictwo genetyczne
Ryzyko genetyczne powtórnego pojawienia się dziecka z zespołem Angelmana u rodzeństwa zależy od defektu molekularnego, jaki tą chorobę wywołał. W większości przypadków ryzyko to nie jest istotnie podwyższone (nie wyższe o więcej niż 1% w stosunku do ryzyka populacyjnego) – sytuacja taka ma miejsce, gdy zespół Angelmana jest spowodowany delecją (najczęstszy przypadek), disomią jedorodzicielską ojcowską, defektem imprintingu (ale nie wynikającym z mutacji tego regionu). W przypadku gdy zespół Angelmana jest spowodowany mutacją w genie UBE3A lub w centrum imprintingu (ale pod warunkiem, że mama jest również nosicielką tej mutacji) – ryzyko to wynosi 50% – taka sytuacja ma miejsce w około 10% przypadków. W rzadkich przypadkach, gdy przyczyną zespołu Angelmana są wady chromosomowe – ryzyko jest zróżnicowane i może sięgać nawet 100% (pojedyncze przypadki). Gdy defektu molekularnego nie udaje się ustalić, a ma to miejsce w 10% do 15% – nie da się ustalić ryzyka genetycznego.