
Produktem genu UBE3A jest białko UBE3A (inna nazwa E6-AP), które jest istotnym elementem szlaku ubikwitynozależnej degradacji białek w proteasomie (przedstawionego poniżej). Szlak ten jest niezwykle istotny dla wszystkich komórek, szczególnie dla komórek nerwowych mózgu. W szlaku tym dochodzi do przyłączenia małego białka ubikwytyny do innych białek, co powoduje ich niszczenie. Ubikiwtyna jest małym białkiem, składa się z 76 aminokwasów, a jej dołączenie do innego białka inicjuje jego destrukcję. Jak to przedstawiono na poniższej rycinie, białka E1 oraz E2 aktywują ubikwitynę (żółta) i przenoszą ją na białko E3. Jest wiele podtypów białka E3, a białko UBE2A jest jednym z nich. UBE3A ma zdolność chemicznego przyłączania ubikwityny do białek docelowych (czerwone). Niezwykle ważne cechą budowy białka UBE3A jest domena HECT, będąca molekularną kieszenią, umożliwiającą zbliżenie się aktywnej ubikwityny do białka docelowego i ich chemiczne związanie. Niektóre białka docelowe dla UBE3A są znane, jednak nie wiadomo dokładnie, które z nich są powiązane z nieprawidłowościami występującymi w zespole Angelmana. Wiadomo, iż UBE3A jest związana z funkcjonowaniem synaps.
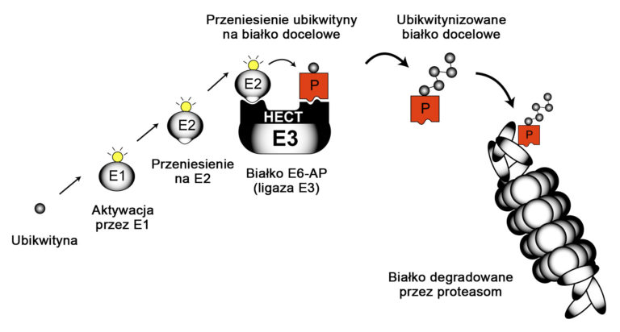
UBE3A i piętno genomowe
Gen UBE3A ulega imprintingowi, czyli piętnowaniu genomowemu w komórkach nerwowych mózgu. Oznacza to, że gen UBE3A, znajdujący się na chromosomie 15 odziedziczonym od ojca jest prawie całkowicie nieaktywny w wielu rejonach mózgu, podczas gdy gen odziedziczony od matki pozostaje w pełni funkcjonalny. Neurony mózgu działają prawidłowo nawet gdy posiadają tylko jedną aktywną kopię genu UBE3A. Delecje będące przyczyną Zespołu Angelmana, dotyczą tylko chromosomu 15 pochodzenia matczynego. W związku z tym, że UBE3A jest aktywny tylko na chromosomie matczynym, delecje takie usuwają jedyną aktywną kopię tego genu. Utrata genów, które są aktywne na chromosomie 15 pochodzenia ojcowskiego, powoduje inny zespół zaburzeń rozwojowych – zespół Pradera-Williego (PWS). W patogenezie PWS również uczestniczą geny ulegające imprintingowi, leżące blisko genu UBE3A, jednak sam wspomniany gen nie bierze udziału w patogenezie tego zespołu. Zespoły AS i PWS stanowią dość unikalną parę zespołów, inne choroby genetyczne związane z piętnowaniem genomowym nie wykazują takiego efektu.
Zrozumienie terminu imprinting, czyli piętnowanie genomowe może nastręczać trudności. Aby geny ulegające piętnowaniu mogły być prawidłowo dziedziczone i aktywne na chromosomie, pochodzącym od właściwego jednego z dwójki rodziców (tak jak u osób zdrowych) musi istnieć mechanizm „czyszczenia” piętna genomowego. Przykładowo, gdy prawidłowy mężczyzna produkuje plemniki, niezależnie od tego, czy przekazuje chromosom 15 pochodzący od swojej matki, czy tez ojca, będzie on piętnowany jako „ojcowski”, a co za tym idzie, gen UBE3A będzie nieaktywny. Odwrotne zjawisko zajdzie u kobiety, u której wszystkie komórki jajowe będą posiadały gen UBE3A w formie aktywnej. Geny piętnowane posiadają zdolność do wymazywania i ponownego nakładania piętna genomowego.
Poniższy rodowód przedstawia jak piętno genomowe może powodować ponowne występowanie potomstwa z Zespołem Angelmana u dość odległych krewnych. Gdy mutacja w genie UBE3A dziedziczy się w rodzinie, osoby, które ją odziedziczyły mogą mieć Zespół Angelmana, ale mogą też być całkowicie zdrowe. Odziedziczenie mutacji genu UBE3A od ojca (drugie pokolenie na rodowodzie) nie powoduje wykrywalnych efektów bezpośrednio u jego dzieci, gdyż ojciec przekazuje nieaktywny gen UBE3A. Obecność mutacji w tym genie u dzieci nie ma dla nich znaczenia, gdyż, każde z nich odziedziczyło prawidłowy chromosom 15 (zawierający prawidłowy aktywny gen UBE3A) od swojej matki. Jednak gdy córka (z pokolenia 2 na przedstawionym rodowodzie) przekaże mutację genu UBE3A swoim dzieciom, będą one miały Zespół Angelmana, gdyż jej dzieci otrzymają nieaktywny gen UBE3A od swojego ojca – nie posiadają więc one aktywnego genu UBE3A. Taki sam typ dziedziczenia może występować w rodzinach, w których doszło do defektu centrum imprintingu (patrz niżej).
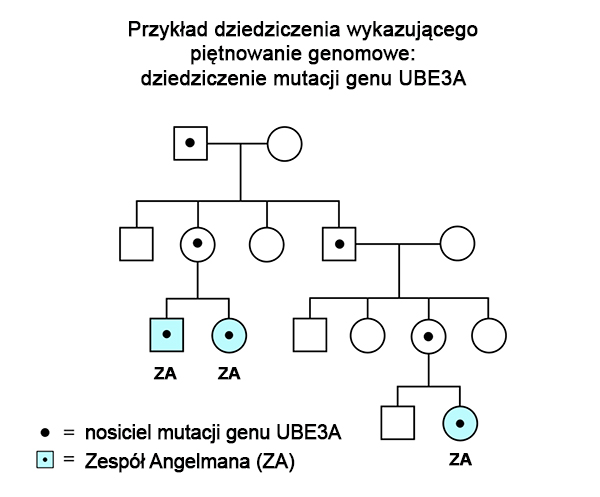
Na szczęście większość pacjentów z zespołem Angelmana nabywa tej choroby poprzez obecność niedziedzicznej, spontanicznej mutacji. Z tą sytuacją mamy do czynienia w przypadkach częstych, dużych delecji, w których piętnowanie w rodowodzie nie jest obserwowane.
Mechanizm genetyczny i ciężkość objawów
Najogólniej rzecz biorąc, wszystkie mechanizmy molekularne, powodujące Zespół Angelmana prowadzą do dość jednolitego obrazu klinicznego, obejmującego niepełnosprawność intelektualną od ciężkiej po głęboką, charakterystyczne zaburzenia zachowania oraz zahamowanie rozwoju mowy. Istnieją jednak pewnie odrębności, korelujące z określonym genotypem:
1. Delecje powodują najcięższą prezentację kliniczną obejmującą małogłowie, napady padaczkowe, hipopigmentację, zaburzenia ruchowe (np. ataksję, obniżone napięcie mięśniowe, trudności w karmieniu) oraz zaburzenia poznawcze i rozwoju mowy.
2. Disomie jednorodzicielskie oraz mutacje centrum imprintingu skutkują łagodniejszymi zaburzeniami rozwojowymi (np. mniej prawdopodobne jest wystąpienie małogłowia), mniejszymi zaburzeniami ruchowymi i powodują mniejszą częstość (ale nie brak) padaczki.
3. Defekty imprintingu wykazują tendencję do najlepszego rozwoju w zakresie zdolności intelektualnych, mowy oraz ruchu w stosunku do pozostałych podgrup. Najbardziej zaawansowany rozwój mowy występuje u pacjentów z defektem imprintingu występującego w mozaice z brakiem defektu (około 20% pacjentów). Osoby te mogą mieć zasób słów do 50-60 słów i używać prostych zdań.
4. Efekt mutacji genu UBE3A jest najczęściej pośredni między delecjami a defektami imprintingu, w zakresie występowania małogłowia, padaczki, zaburzeń motorycznych i rozwoju mowy. Niektóre osoby z mutacją w genie UBE3A wykazują szczególnie łagodny fenotyp, prawdopodobnie dlatego, że występująca u nich mutacja (a dokładniej jej pozycja w genie UBE3A) powoduje mniejsze zaburzenia funkcjonalne.
Diagnostyka
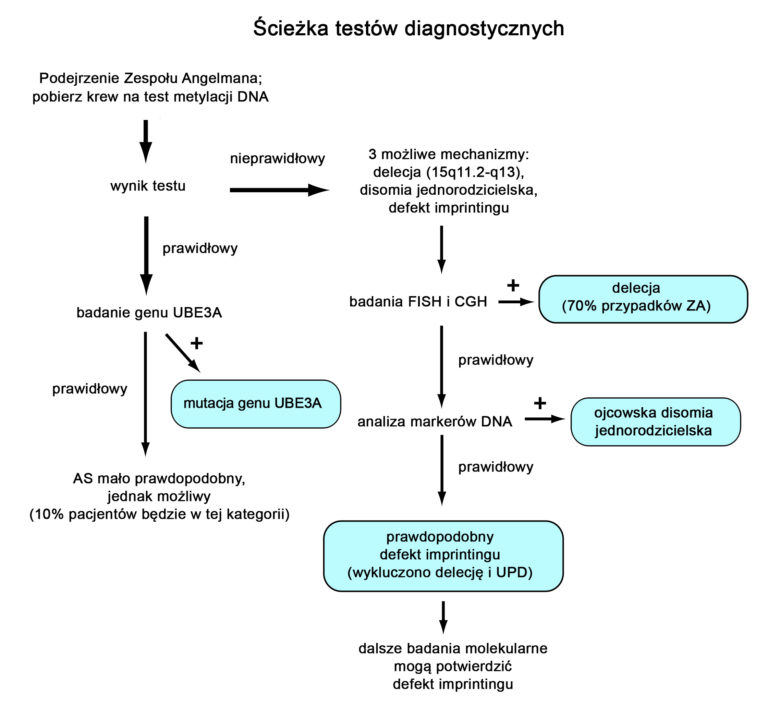
Diagnostyka laboratoryjna Zespołu Angelmana może być złożona. Schemat diagnostyki laboratoryjnej w u osoby, u której podejrzewany jest ZA (rycina powyżej) w pierwszej kolejności obejmuje wykonanie testu metylacji regionu ZA/PWS. Test metylacji jest pozytywny w trzech przypadkach – dużych powszechnych delecji, disomii jednorodzicielskiej oraz defektach centrum imprintingu. Jeżeli test metylacji jest pozytywny, konieczne są dodatkowe badania, aby określić rodzaj defektu genetycznegi. W takich sytuacjach zwykle rozpoczyna się od wykonania badania FISH (fluorescencyjna hybrydyzacja in situ) celu identyfikacji częstych delecji w regionie 15q11.2-13 (istnieją również inne metody mogące potwierdzić tą delecję – np. Porównawcza hybrydyzacja genomowa CGH). Jeżeli test FISH wykluczy delecję, kolejnym krokiem są dodatkowe badania z krwi rodziców, potwierdzające lub wykluczające UPD. Uważa się, że, jeżeli testy FISH otaz w kierunku UPD są prawidłowe, u pacjenta występuje defekt centrum imprintingu. Możliwe jest wykonanie dodatkowych badań, wykazujących delecję DNA w centrum imprintingu (jedak są one dostępne tylko w niewielkiej liczbie laboratoriów). Jeżeli test metylacji da prawidłowy (negatywny) wynik, można wykonywać badanie genu UBE3A celem wykazania mutacji.
Poradnictwo genetyczne i ryzyko genetyczne
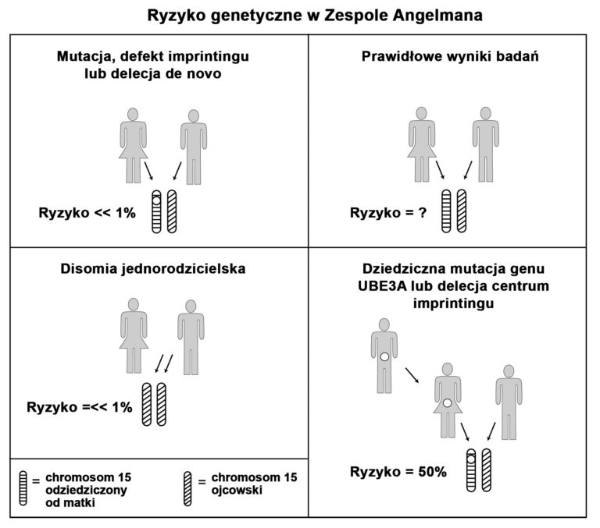
Poniższe aspekty muszą być brane pod uwagę, podczas analizy ryzyka genetycznego w AS. Z uwagi na stopień skomplikowania oceny ryzyka, wskazana jest porada genetyczna udzielona przez genetyka klinicznego.