
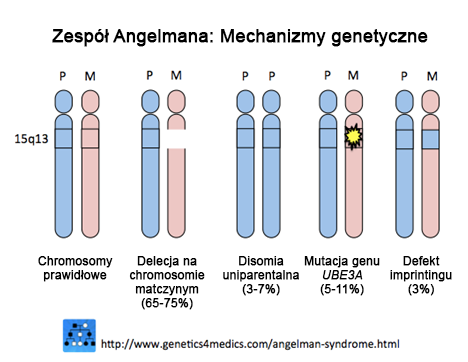
Objawy Zespołu Angelmana wynikają z zakłócenia funkcji genu UBE3A na matczynym allelu chromosomu 15. Każdy człowiek posiada 2 allele (kopie genu), jeden od mamy (matczyny) i jeden od taty (ojcowski). Aby organizm człowieka funkcjonował prawidłowo wystarczy aby tylko jeden allel był aktywny i działał prawidłowo. W ZA ojcowski allel na „szczycie” segmentu kodującego gen UBE3A ma „znak STOP” i dlatego nie działa, więc jedyną nadzieją na funkcjonowanie genu UBE3A jest jego prawidłowe odczytywanie na allelu matczynym. Jeśli na allelu matczynym:
wówczas niedobory, które organizm odczuwa z powodu braku genu UBE3A powodują powstanie choroby nazwanej Zespołem Angelmana.
Cechy genu UBE3A:
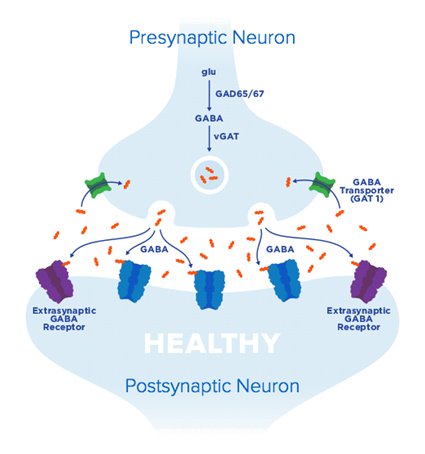
Geny tworzą białka. Białka to neurotransmitery (przekaźniki), które albo hamują przewodnictwo nerwowe (inhibition) albo stymulują to przewodnictwo (excitation). Gen UBE3A tworzy białka, które zajmują się głównie hamowaniem przewodnictwa. Hamowanie to w zdrowym organizmie umożliwia utrzymanie nerwów, mięśni a co za tym idzie całego ciała w stanie idealnej równowagi, spokoju i ciszy. Hamowanie to pozwala też usunąć nadmiar bodźców, szczególnie nadmierne zewnętrzne pobudzenia, tzw. „background noise” (szum otoczenia), które docierają do mózgu. Jest też właściwie krytyczne dla centralnego układu nerwowego w celu rozróżnienia pożądanych sygnałów płynących do mózgu od sygnałów zakłócających, tzw. hałasu. Dzięki temu jesteśmy w stanie się uczyć, skupić, zapamiętywać, spać, odpowiednio się zachowywać a nawet utrzymywać równowagę. Jeśli do mózgu dociera zbyt wiele bodźców a jednocześnie zdolność hamowania tych bodźców (tonic inhibition) jest zaburzona, wówczas ciągłe pobudzenie nerwów skutkuje m.in. napadami padaczkowymi, zaburzeniami koordynacji i motoryki, zaburzeniami snu, zaburzeniami koncentracji a nawet lękiem i niepokojem. To właśnie wyjaśnia kliniczne objawy Zespołu Angelmana, w którym brakuje genu UBE3A, a zatem brakuje też dostatecznego hamowania bodźców.
A dokładniej:Gen UBE3A dostarcza instrukcji do tworzenia białka UBE3A. Białko to jest enzymem „kierownikiem” – pomaga rozpoznawać inne białka w mózgu i kierować nimi – dostarcza informacji których białek potrzebujemy w danej chwili więcej a których mniej. Dlatego w przypadku braku białka UBE3A w mózgu powstaje choas – tworzą się miejsca z nadmiarem lub niedoborem odpowiednich białek. ”Oczyszczające” działanie białka UBE3A jest konieczne do utrzymania prawidłowego funkcjonowania komórek. Gdy zaburzona jest zdolność usuwania białek wówczas dochodzi do nadprodukcji zbędnych materiałów w/wokół neuronów czy rdzenia kręgowego. W Zespole Angelmana przekłada się to bezpośrednio na nadprodukcje receptorów GAT1. Są to receptory, które wychwytują neurotransmitery GABA. O dzieciach z ZA mówimy, że są ‘GABA deficiency” czyli brakuje im przekaźników GABA. Neurotransmitery GABA :
pozwalają racjonalnie planować i wykonywać z sensem to ,co zaplanowaliśmy.
Na przykład:
Jeśli planujemy postawić krok to GABA ‘mówią’ – „aby zrobić krok prawą nogą musisz pobudzić tą prawą nogę a zahamować lewą, spiąć prawą, rozluźnić lewą”. Takie pobudzenie i zahamowanie dają odpowiedni balans. Jeśli jest nadmiar pobudzania to mówimy o utracie zdolności tonicznego hamowania. Jak wspomniałam już wyżej UTRATA TONICZNEGO HAMOWANIA odpowiada za: 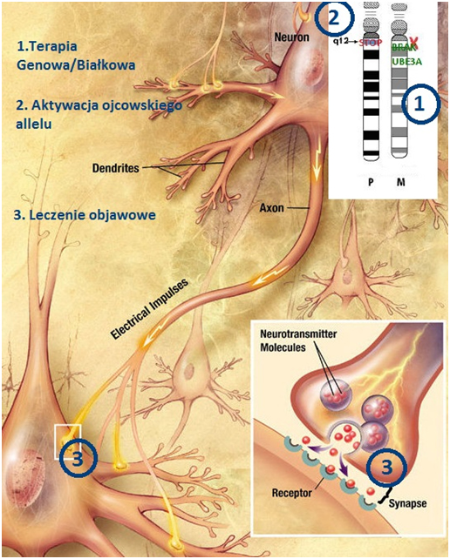
Terapia genowa A. W chwili obecnej trwają zaawansowane badania kliniczne w innych chorobach neurogenetycznych:
A. Polega na zastopowaniu znaku STOP który jest na ojcowskim allelu chromosomu 15 (tzw. STOP the STOP). Wykorzystywana jest tu m.in cząsteczka nazwaną „małą molekułą” złożoną z oligonukleotydów ASO. Molekuła ta ma możliwość przyłączania się do znaku STOP na ojcowskim allelu i ma za zadania zastopować STOP, czyli uruchomić nieczynny gen. B. W roku 2015 w NATURE dr Art Beaudet opublikował duży artykuł na temat zakończonych sukcesem badań u ‘myszy Angelmana’, udało się włączyć ojcowski allel C. Trwa III faza badań klinicznych u osób z SMA, są publikacje na temat pierwszych wyleczonych osób
A. Gaboxadol (OV101) – mała molekuła pochodząca od grzyba Amanita fuscaria. Jest to lek, który działa na synapsy czyli połączenia między nerwami. Zwiększa on hamowanie toniczne poprzez zwiększenie ilości neurotransmitera GABA. W ZA receptory GAT1, które niszczą GABA pracują zbyt aktywnie i „zjadają” neurotransmitery GABA, natomiast receptory które powinny wychwytywać GABA pracują zbyt leniwie. Gaboxadol ma za zadanie bardziej uaktywnić receptory dla GABA, jednocześnie zwiększając ilość krążącego neurotransmitera GABA, którego większa ilość i lepsze wychwytywanie wpłyną na lepsze hamowanie bodźców.
Cechy leku: – tabletki doustne – minimalne efekty uboczne, np. senność, ból głowy, zawroty głowy, nudności, wymioty – lek był już testowany na grupie 3000 pacjentów jako lek nasenny, z różnych powodów nie został jednak zarejestrowany przez firmę – lek był testowany na „myszach Angelmana”, z pozytywnym skutkiem głównie w zakresie motoryki
B. Estry ketonowe – podawanie suplementów zawierających egzogenne ciała ketonowe w celu wprowadzenia organizmu w stan ketozy.
Zrealizować marzenia…
Aby wprowadzić na rynek pojedynczy nowy lek lub nową terapię leczniczą potrzeba lat badań, wysiłku tysięcy naukowców i lekarzy i jak szacują Amerykanie ok 2 biliardów dolarów… Z wielu potencjalnych preparatów leczniczych po latach badań wyłania się zazwyczaj jeden konkretny lek, który po akceptacji FDA w USA (Amerykańska Agencja ds. Żywności i Leków) lub EMA w Europie (Europejska Agencja Leków) wchodzi na rynek. Faza podstawowych badań nad lekiem, odkrycia leku i badań przedklinicznych na zwierzętach trwa zazwyczaj 3-6 lat. Faza badań klinicznych na ludziach 2-7 lat. Faza ostatecznego monitorowania przez FDA i dopuszczenia leku do obrotu ok 0,5-2 lat.
LEK OV101
Ze wszystkich wymienionych wcześniej terapii najbardziej zaawansowane prace w Zespole Angelmana dotyczą leku OV101. I mamy wreszcie kolejny krok do przodu: 6 sierpnia 2018 roku amerykańska firma Ovid zaprezentowała światu pierwsze wyniki badania STARS, na które czekaliśmy od stycznia. Są to pierwsze informacje o leku OV101 od czasu kiedy na konferencji w Phoenix w 2017 r. ogłoszono nabór do 2 fazy badania klinicznego STARS na dorosłych pacjentach z ZA:
Ovid Therapeutics Inc., biofarmaceutyczna firma ukierunkowana na opracowanie leków, które mają zmieniać życie osób z chorobami rzadkimi ogłosiła, że faza 2 badania STARS nad lekiem OV101 osiągnęła swój podstawowy założony punkt końcowy w zakresie bezpieczeństwa i tolerancji. Badany lek wykazał korzystny profil bezpieczeństwa i był dobrze tolerowany wśród dorosłych i młodzieży z zespołem Angelmana. OV101 jest jedynym selektywnym zewnątrzsynaptycznym agonistą receptora GABAa, który pośredniczy w zjawisku tonicznego hamowania, zjawisku, które odpowiada za patomechanizm Zespołu Angelmana. Jeśli OV101 zostanie dopuszczony do rejestracji będzie pierwszym lekiem skierowanym bezpośrednio na czynniki leżące u podłoża Zespołu Angelmana, czyli upośledzone hamowanie toniczne powstające w wyniku zakłócenia pracy genu UBE3A.
Faza 2 międzynarodowego badania STARS była pierwszym komercyjnym, randomizowanym, podwójnie ślepym klinicznym badaniem dedykowanym zespołowi Angelmana, z kontrolną grupą placebo. Po raz pierwszy w historii stworzono solidne badanie kliniczne, które pozwala oszacować konkretne punkty końcowe, a tylko takie badanie może utorować drogę do rejestracji leku. W badaniu losowo przyporządkowano 88 pacjentów do 3 grup: grupa przyjmująca lek OV101 raz dziennie, grupa przyjmująca lek dwa razy dziennie i grupa kontrolna placebo. Przy 12-tygodniowej analizie skuteczności przy użyciu skali CGI-I lek OV101 w porównaniu z placebo wykazał statystycznie istotne korzystne działanie. CGI-I jest skalą często używaną w badaniach klinicznych, która pozwala uchwycić konstelacje różnych objawów klinicznych i pomaga określić wpływ leku zarówno na samych chorych jak i funkcjonowanie rodziny. W badaniu STARS to właśnie skala CGI-I została uznana na nadrzędne narzędzie oceny skuteczności leku. Dalsza analiza objawów obejmowała skale specyficzne do oceny poszczególnych objawów jak np. zachowania, snu czy chodu i w tym zakresie nie uzyskano statystycznie istotnych różnic w porównaniu z placebo, jednak pełna analiza danych nie została jeszcze przedstawiona a badania trwają. Firma Ovid konsultuje obecnie te obserwacje z organami regulującymi rejestracje leków.
Badanie STARS było zaprojektowane tak by dostarczyć jak najwięcej informacji, które pozwolą udoskonalić rozwój OV101 ale też pozwolą określić punkty końcowe, których osiągnięcie jest niezbędne do rejestracji leku. Co istotne – badanie pozwoliło założyć, że przyjmowanie leku jeden raz dziennie będzie wystarczające do osiągnięcia zadowalającego efektu klinicznego. W IV kwartale tego roku Ovid planuje nabór do rozszerzonego otwartego badania ELARA. Będą mogli w nim uczestniczyć pacjenci, którzy już wcześniej uczestniczyli w badaniach klinicznych prowadzonych przez Ovid. Opis badania STARSSTARS było 12-tygodniowym badaniem fazy 2 z podwójnie ślepą próbą i z kontrolą placebo. Dotyczyło 88 pacjentów ( 66 dorosłych i 22 młodzieży) w wieku 13-49 lat, którzy zostali przydzieleni do 13 ośrodków badawczych w Stanach i Izraelu. W badaniu podzielono pacjentów na 3 grupy:
– przyjmujący lek raz dziennie wieczorem w dawce 15mg (QD) – przyjmujący lek dwa razy dziennie 10mg rano i 15mg wieczorem (BID) – przyjmujący placebo Z 88 potencjalnych pacjentów ostatecznie w badaniu wzięło udział 87 osób (średnia wieku 22,6) i każda z nich przyjęła chociaż jedną dawkę OV101, co pozwoliło ująć ją w ocenie założonych punktów końcowych. Podstawowym założeniem końcowym badania była ocena bezpieczeństwa i tolerancji OV101 w porównaniu z placebo. Dodatkowo badanie pozwoliło przeanalizować użyteczność leku w zakresie ogólnego „klinicznego wrażenia”, niepożądanych zachowań, zaburzeń snu a także małej i dużej motoryki. Założenie podstawowe – bezpieczeństwo i tolerancjaBadanie spełniło oczekiwane założenia ukazując podobną ilość efektów ubocznych zarówno podczas przyjmowania leku jak i przyjmowania placebo, z czego większość efektów ubocznych miało nasilenie umiarkowane. OV101 wykazał korzystny profil ryzyka i był dobrze tolerowany przez cały 12-tygodniowy czas trwania badania.
Najczęstszymi zanotowanymi objawami ubocznymi stosowania OV101 były : wymioty, senność, drażliwość, agresja i gorączka. Najczęstsze efekty uboczne:| Efekt uboczny | Placebo (n=29) | OV101 QD (n=29) | OV101 BID (n=29) |
| wymioty | 9 (31%) | 5 (17,2%) | 1 (3,4%) |
| senność | 5 (17,2%) | 5 (17,2%) | 2 (6,9%) |
| drażliwość | 4 (13,8%) | 3 (10,3%) | 3 (10,3%) |
| agresja | 5 (17,2%) | 4 (13,8%) | 1 (3,4%) |
| gorączka | 2 (6,9%) | 7 (24,1%) | 2 (6,9%) |
| Efekt uboczny | Placebo (n=29) | OV101 QD (n=29) | OV101 BID (n=29) |
| gorączka | 2 (6,9%) | 7 (24,1%) | 1 (3,4%) |
| wysypka | 1 (3,4%) | 3 (10,3%) | 2 (6,9%) |
| napady padaczkowe | 0 | 2 (6,9%) | 3 (10,3%) |
| moczenie | 0 | 2 (6,9%) | 1 (3,4%) |
| napady miokloniczne | 0 | 1 (3,4%) | 2 (6,9%) |
Poważne efekty uboczne w postaci napadów padaczkowych zostały zanotowane w 2 przypadkach : jeden pacjent przyjmujący OV101 raz dziennie, ale napad ten został w badaniu uznany za niezwiązany z badaniem, drugi pacjent przyjmujący OV101 dwa razy dziennie i ten napad został określony jako potencjalnie związany z badaniem.
Zaprzestanie kontynuacji udziału w badaniu z powodu efektów ubocznych było marginalne. Z badania zrezygnował 1 pacjent z grupy placebo (zgłaszano drażliwość) i 3 pacjentów z grupy BID (zgłaszano u pacjenta nr 1 mioklonie, nr 2 napady padaczkowe, nr 3 drażliwość/agresję/zaburzenia snu). Żaden pacjent z grupy QD nie zrezygnował z badania. Skuteczność punktów końcowychW 12 tygodniu leczenia oceniono skuteczność terapii przy użyciu skali CGI-I. Wykazano statystycznie istotną poprawę u pacjentów przyjmujących OV101 – poprawa u 38 pacjentów (66,7%) w porównaniu z placebo – poprawa u 11 pacjentów (39,3%). Najlepsze efekty uzyskano u pacjentów przyjmujących lek raz dziennie. Zauważono również znacznie lepszą odpowiedź młodszych pacjentów w porównaniu do starszych. Poprawa dotyczyła 83% pacjentów w wieku 13-17 , 83% pacjentów w wieku 18-24 oraz 70% pacjentów w wieku 25-49.
Pełna prezentacja wyników badania STARS: Plik Ovid STARS Clinic Trial Więcej informacji pod adresem http://www.angelmanstudy.com/Równocześnie trwają prace A-BOM konsorcjum nad stworzeniem wiarygodnych, miarodajnych testów i biomarkerów, które pozwolą monitorować postępy i oceniać skuteczność terapii. Jest to w tej chwili kluczowy, najważniejszy aspekt ponieważ FDA zaakceptuje do użytku tylko taki lek, który będzie miał znaczący wpływ na poprawę jakości życia pacjentów, ocenianą przez parametry uwzględnione przez firmę już na wstępie badania. Inaczej mówiąc, OVID musi mieć konkretne założenia co do działania leku, poparte konkretnymi parametrami, konkretnymi testami czynnościowymi i w trakcje trwania badania udowodnić, że lek działa właśnie tak jak zakładano.
TERAPIA GENOWA
W przypadku terapii genowej firmy koncentrują się na ostatniej fazie badań przedklinicznych, czyli testach bezpieczeństwa na dużych zwierzętach (szczurach i świniach), co jest niezbędne aby FDA dopuściła terapię do I fazy badań klinicznych.
Ponadto trwają poszukiwania ‘idealnego wektora wirusowego’. Takiego, który będzie dawał najmniejszą własną odpowiedź immunologiczną organizmu, umożliwi najlepszą dystrybucję i zapewni najlepszą drogę podania.
AKTYWACJA OJCOWSKIEGO ALLELU
Prócz testów bezpieczeństwa na dużych zwierzętach w chwili obecnej trwają głównie prace nad poznaniem sekwencji znaku STOP na ojcowskim allelu. Sekwencja STOPu u myszy jest znana i udało się ją zastopować ale nie ma nadal do końca pewności czy sekwencja STOPu u ludzi jest podobna i do końca zbadana.